
Pre-training genomic language models using AWS HealthOmics and Amazon SageMaker
Genomic language models are a new and exciting field in the application of large language models to challenges in genomics. In this blog post and open source project, we show you how you can pre-train a genomics language model, HyenaDNA, using your genomic data in the AWS Cloud. Here, we use AWS HealthOmics storage as a convenient and cost-effective omic data store and Amazon Sagemaker as a fully managed machine learning (ML) service to train and deploy the model.
Genomic language models
Genomic language models represent a new approach in the field of genomics, offering a way to understand the language of DNA. These models use the transformer architecture, a type of natural language processing (NLP), to interpret the vast amount of genomic information available, allowing researchers and scientists to extract meaningful insights more accurately than with existing in silico approaches and more cost-effectively than with existing in situ techniques.
By bridging the gap between raw genetic data and actionable knowledge, genomic language models hold immense promise for various industries and research areas, including whole-genome analysis, delivered care, pharmaceuticals, and agriculture. They facilitate the discovery of novel gene functions, the identification of disease-causing mutations, and the development of personalized treatment strategies, ultimately driving innovation and advancement in genomics-driven fields. The ability to effectively analyze and interpret genomic data at scale is the key to precision medicine, agricultural optimization, and biotechnological breakthroughs, making genomic language models a possible new foundational technology in these industries.
Some of the pioneering genomic language models include
- DNABERT which was one of the first attempts to use the transformer architecture to learn the language of DNA. DNABERT used a Bidirectional Encoder Representations from Transformers (BERT, encoder-only) architecture pre-trained on a human reference genome and showed promising results on downstream supervised tasks.
- Nucleotide transformer has a similar architecture to DNABERT and showed that pre-training on more data and increasing the context window size improves the model’s accuracy on downstream tasks.
- HyenaDNA uses the transformer architecture, like other genomic models, except that it replaces each self-attention layer with a Hyena operator. This widens the context window to allow processing of up to 1 million tokens, substantially more than prior models, allowing it to learn longer-range interactions in DNA.
In our exploration of cutting-edge models that push the boundaries of genetic sequence analysis, we focused on HyenaDNA. Pretrained HyenaDNA models are readily accessible on Hugging Face. This availability facilitates easy integration into existing projects or the starting point for new explorations in genetic sequence analysis.
AWS HealthOmics and sequence stores
AWS HealthOmics is a purpose-built service that helps healthcare and life science organizations and their software partners store, query, and analyze genomic, transcriptomic, and other omics data and then generate insights from that data to improve health and drive deeper biological understanding. It supports large-scale analysis and collaborative research through HealthOmics storage, analytics, and workflow capabilities.
With HealthOmics storage, a managed omics focused findable accessible, interoperable, and reusable (FAIR) data store, users can cost effectively store, organize, share, and access petabytes of bioinformatics data efficiently at a low cost per gigabase. HealthOmics sequence stores deliver cost savings through automatic tiering and compression of files based on usage, enable sharing and findability through the biologically focused metadata and provenance tracking, and provide instant access to frequently used data through low latency Amazon Simple Storage Service (Amazon S3) compatible APIs or HealthOmics native APIs. All of this is delivered by HealthOmics, removing the burden of managing compression, tiering, metadata, and file organization from customers.
Amazon SageMaker
Amazon SageMaker is a fully managed ML service offered by AWS, designed to reduce the time and cost associated with training and tuning ML models at scale.
With SageMaker Training, a managed batch ML compute service, users can efficiently train models without having to manage the underlying infrastructure. SageMaker notably supports popular deep learning frameworks, including PyTorch, which is integral to the solutions provided here.
SageMaker also provides a broad selection of ML infrastructure and model deployment options to help meet all your ML inference needs.
Solution overview
In this blog post we address pre-training a genomic language model on an assembled genome. This genomic data could be either public (for example, GenBank) or could be your own proprietary data. The following diagram illustrates the workflow:
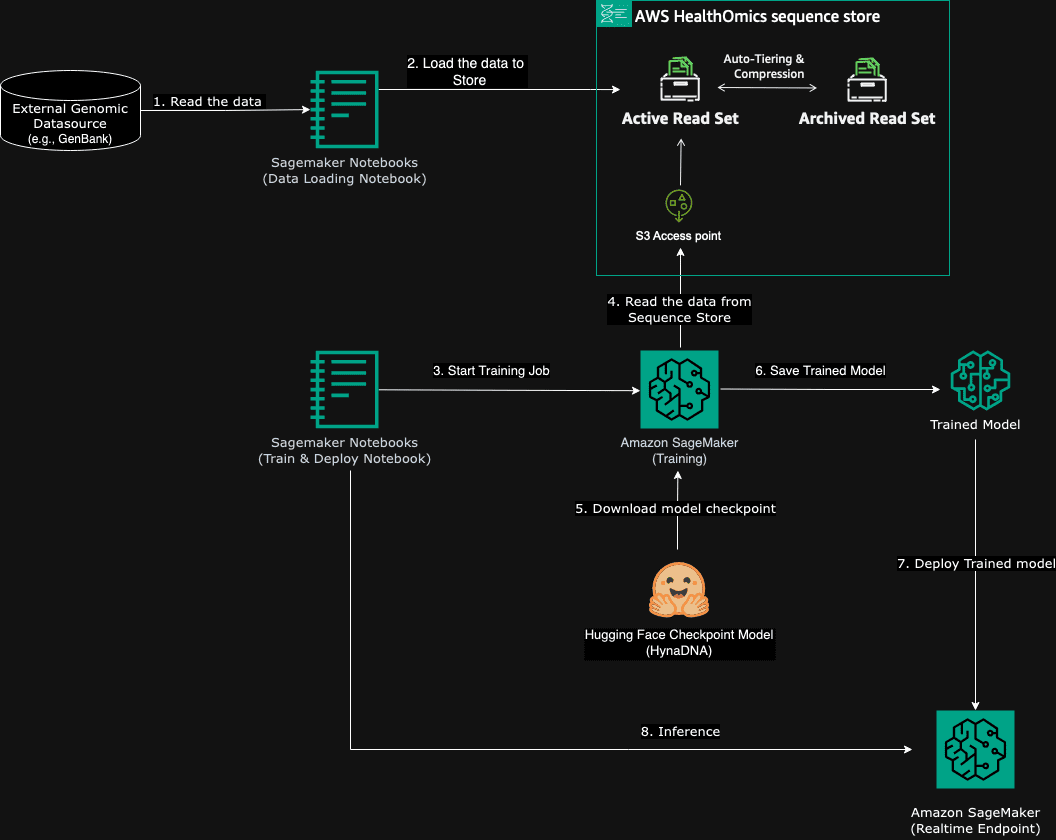
- We start with genomic data. For the purposes of this blog post, we’re using a public non-reference Mouse genome from GenBank. The dataset is part of The Mouse Genomes Project and represents a consensus genome sequence of inbred mouse strains. This type of genomic data could readily be interchanged with proprietary datasets that you might be working with in your research.
- We use a SageMaker notebook to process the genomic files and to import these into a HealthOmics sequence store.
- A second SageMaker notebook is used to start the training job on SageMaker.
- Inside the managed training job in the SageMaker environment, the training job first downloads the mouse genome using the S3 URI supplied by HealthOmics.
- Then the training job retrieves the checkpoint weights of the HyenaDNA model from Huggingface. These weights are pretrained on the human reference genome. This pretraining allows the model to understand and predict genomic sequences, providing a comprehensive baseline for further specialized training on a variety of genomic tasks.
- Using these resources, the HyenaDNA model is trained, where it uses the mouse genome to refine its parameters. After pre-training is complete and validation results are satisfactory, the trained model is saved to Amazon S3.
- Then we deploy that model as a SageMaker real-time inference endpoint.
- Lastly the model is tested against a set of known genome sequences using some inference API calls.
Data preparation and loading into sequence store
The initial step in our machine learning workflow focuses on preparing the data. We start by uploading the genomic sequences into a HealthOmics sequence store. Although FASTA files are the standard format for storing reference sequences, we convert these to FASTQ format. This conversion is carried out to better reflect the format expected to store the assembled data of a sequenced sample.
In the sample Jupyter notebook we show how to download FASTA files from GenBank, convert them into FASTQ files, and then load them into a HealthOmics sequence store. You can skip this step If you already have your own genomic data in a sequence store.
Training on SageMaker
We use PyTorch and Amazon SageMaker script mode to train this model. Script mode’s compatibility with PyTorch was crucial, allowing us to use our existing scripts with minimal modifications. For the training, we extract the training data from the sequence store through the sequence store’s provided S3 URIs. You can, for example, use the boto3 library to obtain this S3 URI.
When you provide this to the SageMaker estimator, the training job takes care of downloading the data from the sequence store through its S3 URI. Following Nguyen et al, we train on chromosomes 2, 4, 6, 8, X, and 14–19; cross-validate on chromosomes 1, 3, 12, and 13; and test on chromosomes 5, 7, and 9–11.
To maximize the training efficiency of our HyenaDNA model, we use distributed data parallel (DDP). DDP is a technique that facilitates the parallel processing of our training tasks across multiple GPUs. To efficiently implement DDP, we used the Hugging Face Accelerate library. Accelerate simplifies running distributed training by abstracting away the complexity typically associated with setting up DDP.
After you have defined your training script, you can configure and submit a SageMaker training job.
First, let’s define the hyperparameters, starting with model_checkpoint. This parameter refers to a HuggingFace model ID for a specific pre-trained model. Notably, the HyenaDNA model lineup includes checkpoints that can handle up to 1 million tokens. However, for demonstration purposes, we are using the hyenadna-small-32k-seqlen-hf model, which has a context window of 32,000 tokens, indicated by the max_length setting. It’s essential to understand that different model IDs and corresponding max_length settings can be selected to use models with smaller or larger context windows, depending on your computational needs and objectives.
The species parameter is set to mouse, specifying the type of organism the genomic training data represents.
Next, define what metrics, especially the training and validation perplexity, to capture from the training logs:
Finally, define a Pytorch estimator and submit a training job that refers to the data location obtained from the HealthOmics sequence store.
Results
In our training cycle for the model, we processed a dataset consisting of one mouse genome with 10,000 entries. The computational resources included a cluster configured with one ml.g5.12xlarge instance, which houses four Nvidia A10G GPUs. The 32k sequence length model, was trained using a batch size of four per GPU (24 gigabit (Gb) of VRAM). With this setup we completed 150 epochs to report the results below.
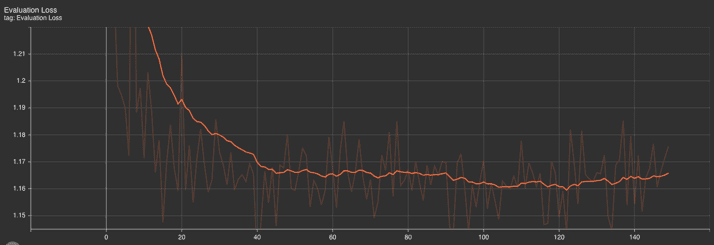
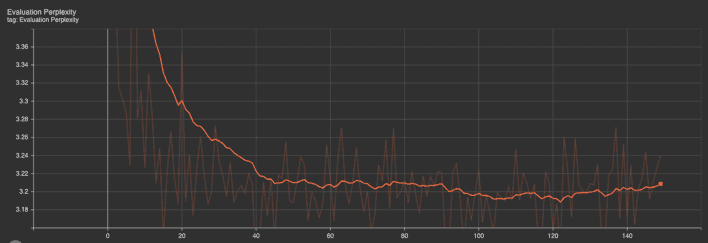
Evaluation metrics: The evaluation perplexity and loss graphs show a downward trend at the outset, which then plateaus. The initial steep decrease indicates that the model rapidly learned from the training data, improving its predictive performance. As training progressed, the rate of improvement slowed, as evidenced by the plateau, which is typical in the later stages of training as the model converges.
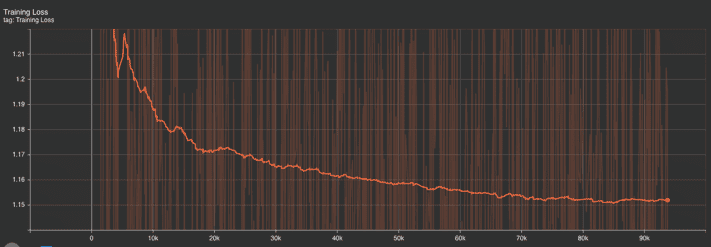
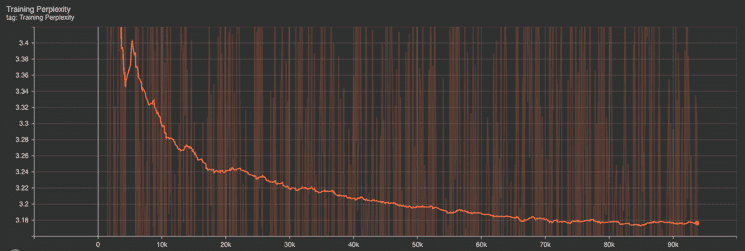
Training Metrics: Similarly, the training perplexity and loss graphs indicate an initial sharp improvement followed by a gradual plateau. This shows that the model effectively learned from the data. The training loss’s slight fluctuations suggest that the model continued to fine-tune its parameters in response to the inherent complexities in the training dataset.
Deployment
Upon the completion of training, we then deployed the model on a SageMaker real-time endpoint. SageMaker real-time endpoints provide an on-demand, scalable way to generate embeddings for genomic sequences.
In our SageMaker real-time endpoint setup, we need to adjust the default configurations to handle large payload sizes, specifically 32k context windows for both requests and responses. Because the default payload size of 6.5 MB isn’t sufficient, we’re increasing it to a little over 50 MB:
By submitting a sequence to the endpoint, users can quickly receive the corresponding embeddings generated by HyenaDNA. These embeddings encapsulate the complex patterns and relationships learned during training, representing the genetic sequences in a form that is conducive to further analysis and predictive modeling. Here is an example of how to invoke the model.
When you submit a sample genomic sequence to the model, it returns the embeddings of that sequence:
Conclusion
We’ve shown how to pre-train a HyenaDNA model with a 32k context window and to produce embeddings that can be used for downstream predictive tasks. Using the techniques shown here you can also pre-train a HyenaDNA model with context windows of other sizes (for example, 1 million tokens) and on other genomic data (for example, proprietary genomic sequence data).
Pre-training genomic models on large, diverse datasets is a foundational step in preparing them for downstream tasks, such as identifying genetic variants linked to diseases or predicting gene expression levels. In this blog post, you’ve learned how AWS facilitates this pre-training process by providing a scalable and cost-efficient infrastructure through HealthOmics and SageMaker. Looking forward, researchers can use these pre-trained models to fast-track their projects, fine-tuning them with specific datasets to gain deeper insights into genetic research.
To explore further details and try your hand at using these resources, we invite you to visit our GitHub repository. Additionally, We encourage you to learn more by visiting the Amazon SageMaker documentation and the AWS HealthOmics documentation.
About the authors
 Shamika Ariyawansa, serving as a Senior AI/ML Solutions Architect in the Global Healthcare and Life Sciences division at Amazon Web Services (AWS), specializes in Generative AI. He assists customers in integrating Generative AI into their projects, emphasizing the adoption of Large Language Models (LLMs) for healthcare and life sciences domains with a focus on distributed training. Beyond his professional commitments, Shamika passionately pursues skiing and off-roading adventures.
Shamika Ariyawansa, serving as a Senior AI/ML Solutions Architect in the Global Healthcare and Life Sciences division at Amazon Web Services (AWS), specializes in Generative AI. He assists customers in integrating Generative AI into their projects, emphasizing the adoption of Large Language Models (LLMs) for healthcare and life sciences domains with a focus on distributed training. Beyond his professional commitments, Shamika passionately pursues skiing and off-roading adventures.
 Simon Handley, PhD, is a Senior AI/ML Solutions Architect in the Global Healthcare and Life Sciences team at Amazon Web Services. He has more than 25 years experience in biotechnology and machine learning and is passionate about helping customers solve their machine learning and genomic challenges. In his spare time, he enjoys horseback riding and playing ice hockey.
Simon Handley, PhD, is a Senior AI/ML Solutions Architect in the Global Healthcare and Life Sciences team at Amazon Web Services. He has more than 25 years experience in biotechnology and machine learning and is passionate about helping customers solve their machine learning and genomic challenges. In his spare time, he enjoys horseback riding and playing ice hockey.
Leave a Reply